Ivemark syndrome with cor triatriatum, primum ASD, cleft mitrale and pulmonary stenosis
Full Text
Introduction
Ivemark syndrome is a rare anomaly with a reported incidence of 1 in 10000 – 20000 live births. It is characterized by agenesis of spleen, a dextropozed, hypoplastic or lobulated spleen or multiple spleens in association with complex cardiac anomalies.[1,2] These malformations are usually associated with trilobulated or bilobulated lungs and abnormalities of other abdominal organs. Some other anomalies like Howell-Jolly and Heinz bodies in peripheral blood, agenesis of corpus callosum and facial abnormalities were also noted.[3,4]
Here we report a case of Ivemark syndrome, accompanied by polysplenia, accessory gall bladder, dilatation of the intrahepatic ducts, cor triatriatum, primum ASD, pulmonary stenosis, dysmorphic face and mild mental retardation.
Case Report
A 20-year-old woman was admitted to Dr. Sami Ulus Research and training Hospital because of dyspnea and palpitation. Family history was unremarkable. At the time of admission her height was 164 cm and body weight was 49 kg. Physical examination revealed that she had mild cyanosis with facial anomalies including prominent nasal bridge, malformed ears, simple philtrum, narrow forehead, hypotelorism and mild mental retardation (Figure 1).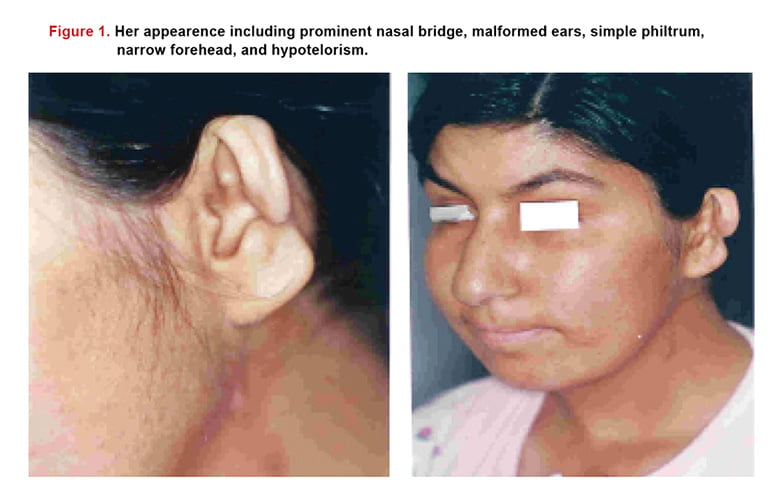
Auscultation revealed augmentation of the S1 sound, and a 20/60 systolic murmur was heard at the left side of the sternum. The heart rate was 72 / min and systolic blood pressure was 90 mm.Hg. The liver was enlarged to 2-3 cm below costal margin. Telecardiogram showed an enlarged heart with a cardiothoracic ratio of 0.6. Transthorasic echocardiography revealed mild pulmonary stenosis with a 45 mm Hg gradient, primum ASD, a membrane in the right atrium and mitral insufficiency (Figure 2). Abdominal ultrasonography indicated polysplenia, accessory gallbladder and dilatation of intrahepatic ducts. Cranial tomography was normal (Figure 3).
She underwent an operation.Cardiopulmonary bypass with bicaval cannulation was directly established from the superior and inferior vena cava. Standart cardiopulmonary bypass with systemic hypothermia at 280 C were used. Cold potassium crystalloid cardioplegia and topical hypothermia were used for myocardial protection.
Then right atriotomi was performed. On exploration it was observed that the right atrium was divided into two portions by a membrane. The membrane was fully resected. The mitral cleft was repaired with 5-0 prolene suture and the primum ASD was closed with a pericardial patch. Pulmonary valvular stenosis was treated by commissurotomy. The post-operative course was uneventfull and she was discharged at 6th postoperative day.
Comment
Ivemark syndrome is a very rare congenital disorder that includes complex cardiac malformations, splenic agenesis and abnormalities of other abdominal organs. Most of the cases are sporadic, but familial occurence also has been described, suggesting autosomal recessive inheritance. The etiology of the syndrome has not been clearly defined, but the teratogenic effect seems to occur between the 30th – 40th days of intrauterin life. A study by Britz-Cunningham indicated that a mutation of the connexin 43 (CX43) gene might also be responsible for Ivemark syndrome.[4]
Ivemark syndrome is a multiple organ syndrome and symmetric liver, malrotation or dilatation of the bowel, duplication of uterus and vagina were observed.[5,6] In our case, in addition to polysplenia, accessory gallbladder, multiple facial anomalies and mild mental retardation were observed. But Ivemark Syndrome’s lung or hematologic signs were absent.
Ivemark syndrome accounts for 1-3 % of all congenital heart defects. The most frequent cardiac malformations are as follows: Persistent atrioventricular canal, patency of ductus arteriosus, atrial septal defect, ventricular septal defect, truncus arteriosus and dextrocardia.[7] The cardiac anomalies can increase the mortality risk and prognosis depends mainly on the degree of malformation of the heart.[8] Due to our knowledge the association of cor triatriatum with Ivemark syndrome has not been reported previously.
References
- Cesko I, Hadju J,Toth T,Papp C,Papp Z.Ivemark Syndrome with asplnia in siblings. J Pediatr 1997; 130:822-824.
- Tkebuchova T, Segesser LK, Lachat M,Genoni M, Bauersfeld U, Turina M.Ivemark syndrome,a case with successful surgical intervention.Scand Cardiovasc 1997;31:173-175.
- Britz-Cunningham SH, Shsh MM, Zuppan CW, Fletcher WH. Mutations of the connexin-43 gap-junction gene in patients with heart malformations and defects laterality.New Engl J Med 1995;332:1323-1329.
- Rodriguez JI, Palacios J, Omenaca F, Lorente M.Polysplenia, caudal deficiency and agenesis of corpus callosum.Am J Med Genet 1991;38:99-102.
- Prabakaran S, Kumaran N, Regunanthan SR, Prasad N, Sridharan S. Spontaneous biliary perforation in a child with features of Ivemark Syndrome. Pediatr Surg 2000;16:109-110.
- Berdat PA, Mohacei P, Althaus U, Carrel T.Succesfull heart transplantation in a patient with Ivemark Syndrome combined with situs inversus,single atrium and ventricle after total cavopulmonary connection.Eur J Cardiovasc Surg 1998;14:631-634.
- Anderson RH. Division of atrial chambers. In: Anderson RH, Baker EJ, Macartney RFJ, Rigby ML,Shinebourn EA, Tynan M (eds.). Pediatric Cardiology 2nd edition. Churchill-Livingston, London; 2002: 931-938.
- Bartram U, Wirbelauer J, Speer CP. Heterotaxy syndrome — asplenia and polysplenia as indicators of visceral malposition and complex congenital heart disease. Biol Neonate 2005;88:278-290.




